
Context
Vascular diseases such as varicose veins and chronic venous insufficiency are common patient presentations in rural medicine. Due to their high prevalence, they are considered to be widely investigated and well understood. Congenital vascular anomalies, however, still represent a challenge in diagnosis and management1. Multiple disorders of the vascular system exist under this umbrella term, including Klippel–Trénaunay syndrome (KTS). KTS is a very rare congenital disorder consisting of a triad of anomalies that typically affect only one limb: capillary malformations, venous malformations and limb hypertrophy1,2. Despite the well-defined and described clinical features of KTS, the epidemiological data are still unsatisfactory.
This report presents the case of a 60-year-old male from a rural community who had previously been treated for, but not diagnosed with, KTS. This case demonstrates how complex congenital vascular malformations can present in rural populations and require physicians in remote areas to have a high index of suspicion for clinical presentations that may harbor more complex disorders than initial assessment suggests. An awareness of the panorama of vascular anomalies will allow for earlier recognition and management of these rare conditions, even in areas where advanced healthcare services or specialists are not available.
Issue
In September 2018, a 60-year-old Caucasian male presented to the surgical clinic at the Charles S. Curtis Memorial Hospital in St Anthony, Newfoundland, with extensive varicosities of the left leg and chronic venous insufficiency. The patient had been recently diagnosed with an advanced osteoarthritis of the right knee, and a total knee replacement was recommended. There was, however, a concern of possible venous thrombosis intraoperatively, so he was referred to the general surgeon for possible phlebectomy prior to the orthopedic surgery.
The patient reported symptoms of fatigue and heaviness in the left leg while walking and moderate swelling at night. He denied having pain or any history of recent trauma. The patient had a longstanding history of varicose veins on the left, affecting both the greater and lesser saphenous distribution. He also experienced multiple episodes of ulceration with severe pain and edema. In 1987, he was hospitalized due to a left popliteal superficial thrombophlebitis. Additionally, he underwent a left-sided saphenofemoral ligation and multiple stab avulsions of his varicosities in 2004. The surgery was uneventful; however, there was a recurrence of the symptomatic varicosities within a few years.
The patient’s past medical history included gastroesophageal reflux disease, chronic obstructive pulmonary disease, non-ST-elevation myocardial infarction and occasional rectal bleeding. Past surgical history included an open cholecystectomy, and a Hartmann`s procedure with subsequent reversal for perforated diverticulitis. No family history of varicosities, port-wine stains or other vascular anomalies were recorded. He had smoked 20 cigarettes a day for 30 years but quit smoking in 2013. His occupation as a fisherman included prolonged periods of standing.
The physical examination confirmed visible varicosities on the left leg. The left foot was warm, with normal capillary refill and strong, palpable pulses. The most striking observation was the port-wine birthmark on the lateral aspect of the left leg, extending from the mid-thigh to the toes. The stain was dark red, raised in some areas, and had sharply demarcated borders, whereas some areas were less well defined. There was no discrepancy in leg length; however, the left leg was larger in circumference than the right. The right leg was normal. Laboratory tests showed normal complete blood count and coagulation parameters, but hyperuricemia (10.3 mmol/L).
A Doppler ultrasound and the ankle-brachial index of the left leg were normal. There were no signs of arteriovenous fistula. The patient underwent a Trendelenburg test in 2004, which showed the incompetence of the saphenofemoral junction. Based on the presence of the triad of a large port-wine stain, extensive varicosities and a larger left leg, KTS was identified in this previously undiagnosed patient.
Due to his symptoms, the patient opted for surgical intervention. The phlebectomy was conducted under spinal anesthesia. At his 6-week postoperative visit, he reported having severe burning pain over the lateral aspect of the left lower leg. This pain resolved slowly over time, but he continued to have reduced sensation to touch over the same area. This also resolved with time. The initial symptoms of swelling, fatigue and heaviness subsided completely after surgery (Fig1).
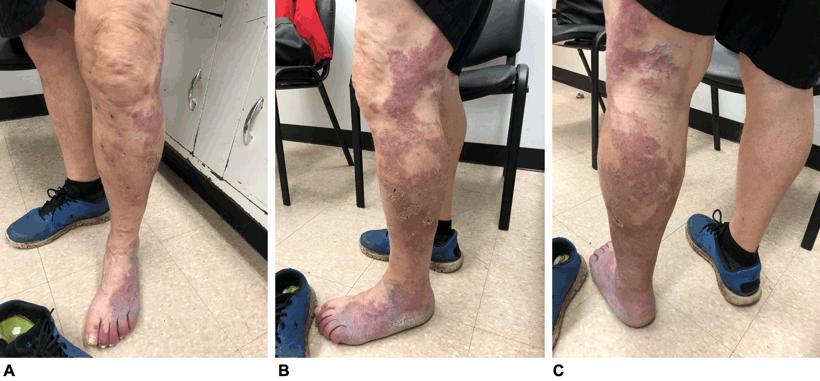 Figure 1: Postoperative images of the patient, showing the large violaceous port-wine stain with sharply demarcated borders and less well defined areas throughout. Multiple black dots on the left leg indicate the areas where the phlebectomy was performed. A. Anterior view; B. lateral view; C. posterior view.
Figure 1: Postoperative images of the patient, showing the large violaceous port-wine stain with sharply demarcated borders and less well defined areas throughout. Multiple black dots on the left leg indicate the areas where the phlebectomy was performed. A. Anterior view; B. lateral view; C. posterior view.
Lessons learned
Vascular disorders are very common and believed to be well known and widely understood. Regardless, congenital anomalies remain an intriguing area in the field of vascular medicine. Missed diagnoses and mistakes in terminology increase the risk of inaccurate evaluation and treatment1,3.
According to the International Society for the Study of Vascular Anomalies’ 2014 classification, vascular anomalies are divided into vascular malformations and vascular tumors. ‘Malformations associated with other anomalies’ represent a further subgroup of vascular malformations to which KTS also belongs. KTS is defined by the triad of capillary and venous malformations as well as limb overgrowth, whereas lymphatic malformation may or may not be present1. Although KTS was first described more than a century ago, the epidemiology and pathophysiology remain to be defined4. KTS, along with other vascular malformations, usually does not regress spontaneously, but can regress with puberty, trauma or infections5,6. KTS is believed to be mostly sporadic, but there is also evidence of familial occurrence7,8. Several studies have found that most KTS patients have somatic phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA) mutations9. These mutations eventually lead to an activation of mammalian target of rapamycin (mTOR) and increased angiogenesis, which explains the therapeutic potential of mTOR inhibitor Sirolimus in KTS. Sirolimus has been shown to be an efficacious and safe alternative in reducing the symptoms and complications of vascular malformations, especially in younger patients5,6,10,11. Recent studies investigated additional inhibitors of the PI3K (phosphoinositide-3-kinase)-Akt-mTOR signaling pathway in KTS. Di Blasio et al have demonstrated in mouse models that a dual therapy with PI3K/mTOR inhibitor BEZ235 or mTOR inhibitor Everolimus could significantly reduce vascular lesions. On the other hand, therapy with Akt inhibitor has been found to be less successful 12. A study by Venot et al revealed that BYL719, a PIK3CA inhibitor, could ameliorate disease symptoms in patients with associated overgrowth syndromes13. In the present case, the patient did not have genomic testing nor did he undergo targeted medical therapy.
The cutaneous capillary malformation in KTS manifests as a port-wine stain (nevus flammeus). As proposed by a recent study, the stains can be stratified into ‘geographic’ or ‘blotchy/segmental’ types14. Geographic stains are described as dark red and sharply demarcated throughout the entire nevus, while segmental stains are rather indistinctly demarcated and light pink. Interestingly, the same study put forward that geographic stains, and not segmental stains, are strongly associated with both lymphatic malformations and other complications in KTS patients14. In this case, the patient had mostly well-defined violaceous stains, although the demarcation in the lateral aspect of the shin was less distinct. The features of both geographic and segmental stains increases the likelihood of a lymphatic malformation.
For physicians in rural areas managing patients with KTS, identification is important in order to manage possible complications. These complications can include coagulopathy, thromboembolism and thrombophlebitis2,15. A previous study revealed that, in a cohort of 75 KTS patients, 29 patients (39%) had had thromboembolic complications. Moreover, some patients had clinically silent thrombosis identified by ultrasonography. As reported by the same study, the KTS patients had significantly higher D-dimer levels and plasmin–antiplasmin complexes, whereas protein C levels were lower compared to controls16. It should be noted that the patient in this case previously had an episode of thrombophlebitis on the affected limb. His coagulation profile was normal during the current evaluation, but he will need further education on identifying symptoms of thromboembolic complications. Symptomatic patients should undergo diagnostic imaging such as Doppler ultrasound17. Low-dose aspirin therapy provided promising results in patients with vascular malformations, reducing the pain and soft tissue swelling18. It may be helpful in case of hypercoagulability. Lifelong anticoagulation is required in patients with deep venous thrombosis or pulmonary embolism16.
Laboratory investigations in this patient revealed only hyperuricemia. Ulrich et al reported hyperuricemia as an independently associated laboratory value in a KTS patient with pulmonary arterial hypertension (PAH)19. Further studies should be undertaken to determine if and how KTS is associated with hyperuricemia and PAH. No studies have yet been performed on this patient to evaluate for pulmonary hypertension, but it is worth considering.
Another possible complication of KTS is bleeding from abnormal vessels. The vascular malformations are often not restricted only to the superficial venous system but may also involve the gastrointestinal and genitourinary tract. This can lead to life-threatening hemorrhages, most frequently from the rectum and bladder20-22. The patient in this case report had a history of occasional rectal bleeding, which points to the possibility of a visceral manifestation of KTS. If clinically significant bleeding occurs, CT or MR angiography may be considered to assess the extent of visceral manifestations20.
Differential diagnosis of KTS includes a number of syndromes with capillary malformations and overgrowth, such as Parkes–Weber syndrome (PWS) and CLOVES syndrome (congenital, lipomatous, overgrowth, vascular malformations, epidermal nevi and skeletal anomalies)23. Most of these syndromes have their own distinct features (Table 1). It can, however, be challenging to distinguish between KTS and PWS1. In this patient’s case, the absence of fast-flow arteriovenous fistulae effectively ruled out PWS and indicated instead a diagnosis of KTS. The fact that the patient was diagnosed at an advanced age clearly demonstrates the need of early diagnostic tools in suspected patients to prevent or decelerate possible complications. An important starting point could be genetic testing in pediatric patients with associated clinical findings. In this regard, there has been growing interest in investigating the genetic background of KTS and related disorders. In recent years, the hypernym ‘PIK3CA-related overgrowth spectrum (PROS)’ was established to classify diseases linked to somatic PIK3CA mutations24. This includes CLOVES syndrome, fibroadipose hyperplasia or overgrowth, megaencephaly-capillary malformation and, more recently, KTS24,25.
Vascular anomalies define a broad scope of disorders ranging from a benign nevus to a syndrome with severe complications. Precise diagnosis of complex vascular anomalies often requires a multidisciplinary approach to ensure appropriate management. Since the presence of medical or surgical subspecialists in rural areas is rare, diagnosis and management fall to the general practitioner. Awareness of the breadth of congenital vascular anomalies and the impact of their possible complications are critical for the rural and remote physician in delivering safe and effective health care. This report seeks to address this challenge and raise awareness of lesser known causes underlying vascular disorders for remote healthcare providers.
Table 1: Differential diagnosis of Klippel–Trénaunay syndrome 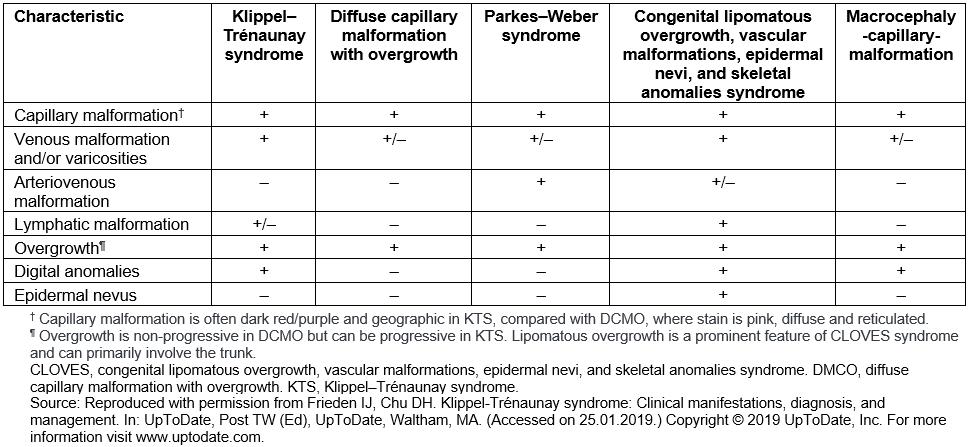
Acknowledgements
The authors thank Kweku Dankwa at Charles S. Curtis Memorial Hospital, Newfoundland, Canada.
References
You might also be interested in:
2016 - Supporting nurses' transition to rural healthcare environments through mentorship